[엠디저널]프레세닐린(Preseniline)은 아밀로이드전구체단백질(APP)과 함께 알츠하이머를 유발하는 가장 대표적인 상염색체 우성상태 유전자로 주로 초로기 가족력 알츠하이머(EOFAD)에 관여하며, 이들 프레세닐린 유전자의 돌연변이는 알츠하이머 뿐만 아니라 여러 종류의 치매와 연관되어 있다.
알츠하이머를 조기치료 및 예방하기 위한 시급한 문제인 조기진단 바이오마커의 개발을 위한 알츠하이머와 유전자의 상관관계를 조사하는 미래 바이오마커 연구회(Prospective Biomarker Studies)는 2012년, 상염색체 우성 알츠하이머 연구를 통해 프레세닐린1(PSEN1)에 E280A 돌연변이를 가진 노인은 질병이 발병하기 약 20년 전부터 뇌에 Aβ가 축적된다는 것을 보고하였다.
프레세닐린(Preseniline, PSEN1과 PSEN2)은 아밀로이드 전구체 단백질(APP)과 함께 알츠하이머를 유발하는 가장 대표적인 상염색체 우성상태 유전자로 주로 초로기 가족력 알츠하이머(Early-Onset Familial Alzheimer’s Disease, EOFAD)에 관여한다. PSEN1과 PSEN2로는 초로기 알츠하이머(Early-Onset Alzheimer’s Disease, EOAD)를 10%정도 밖에 설명하지 못하지만, 대부분이 초로기 알츠하이머(EOAD) 환자에서 프레세닐린 유전자에 대해 기술되어 왔다. 따라서 이러한 기전은 노년기(Late-Onset Alzheimer’s Disease, LOAD) 또는 산발적 알츠하이머(Sporadic Alzheimer’s Disease, SAD)와는 관련이 없는 것으로 알려져 있다.
PSEN1 유전자는 14번 염색체에서, PSEN2는 1번 염색체에서 발견되며, 이들 프레세닐린 유전자의 돌연변이는 알츠하이머뿐만 아니라 여러 종류의 치매와 연관되어 있다. 실제로 PSEN1 유전자의 130개 이상의 돌연변이가 유전학적 연구에서 밝혀졌으며, 치매 발현의 차이는 돌연변이 부위에 따라 다양하다(그림 1).
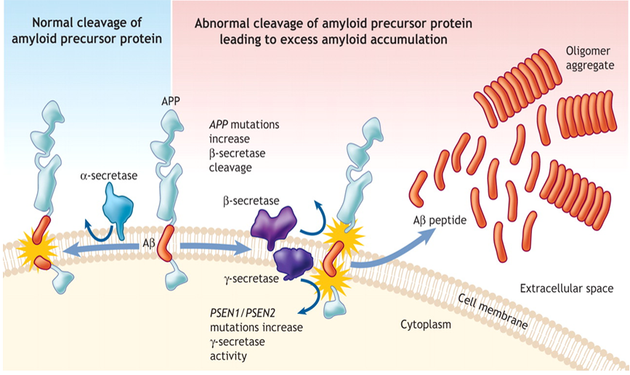
PSEN1, PSEN2 돌연변이 유전자는 Aβ42:Aβ40 비율을 높이며 이 돌연변이가 가족력 알츠하이머를 유발한다. 그렇지만 불완전한 가족력은 알츠하이머의 유전적 위험을 평가하는 데 중요한 장애물이 될 수 있다.
PSEN1 돌연변이 유전자는 Aβ(1-42)와 Aβ(1-40)의 생산을 감소시키는 유리한 γ-세크레타제 활성을 없앤다. 이로 인해 Aβ 침착을 촉진시키는 Aβ(1-42)/Aβ(1-40) 비율을 증가시킬 수 있다. 다시말하면, PSEN1, PSEN2 돌연변이 유전자는 Aβ42:Aβ40 비율을 높이며 이 돌연변이가 가족력 알츠하이머(Familial Alzheimer’s Disease, FAD)를 유발한다. 따라서 FAD에 대한 많은 연구가 γ-세크라타제를 암호화하는 유전자의 돌연변이에 초점을 맞추고 있다(그림 2).
그렇지만 불완전한 가족력은 알츠하이머의 유전적 위험을 평가하는 데 중요한 장애물이 될 수 있다. 개인이나 가족의 유전적 위험을 평가하려면 질병이 진행된 가족의 알츠하이머 진단과 발병 연령에 관하여 가능한 한 많은 정보를 가지고 정확한 가족력을 얻는 것이 중요하지만, 형제가 없거나 형제, 자매의 건강 상태에 관한 정보가 없을 수도 있어 신뢰할 만한 답을 얻는 것은 생각만큼 쉽지 않다.
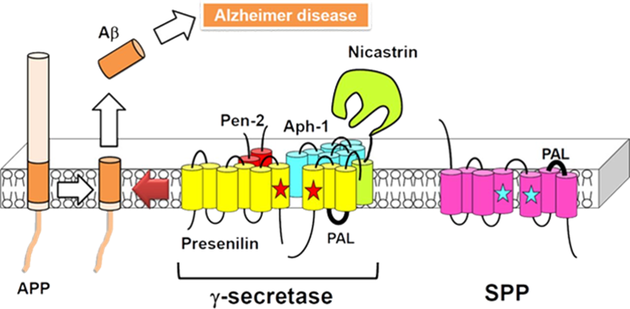
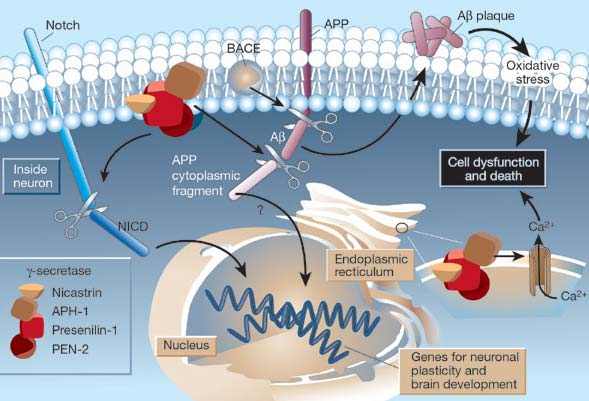
PSEN1과 PSEN2는 γ-세크레타제의 핵심 구성요소로서 원형질막 내에서 β-세크레타제 분해에 의해 생성된 APP의 C-말단 단편을 절단하여 Aβ를 방출한다. 포유류의 γ-세크레타제의 완전한 구조는 아직 알려지지 않았으나 현재까지 γ-세크레타제는 Presenilin1(PSEN1), Nicastrin, Anterior pharynx defective-1(Aph1), Presenilin enhancer-2(PSEN2)의 복합체로 알려져 있다(그림 3).
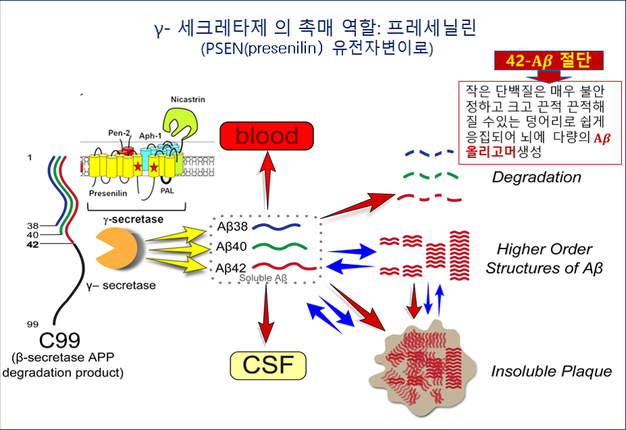
독성이 있는 Aβ(1-42)로 잘리는 것은 γ-세크레타제의 촉매인 PSEN 유전자 때문으로 알츠하이머 환자의 뇌에서 γ-세크레타제의 구조 내에 PSEN2 유전자변이가 발견되며, 42번 β-아밀로이드로 절단된 Aβ(1-42)가 혈액과 뇌척수액(CSF)에서 측정된다(그림 4). 또한 PSEN1 돌연변이 또한 42번 부위에서 γ-세크레타제 분열을 증가시키며, 두 경우 모두 과량의 독성 Aβ(1-42) 펩타이드 생성이 일어난다.
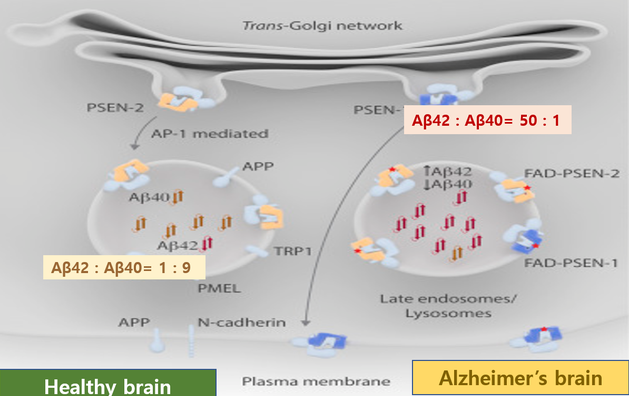
PSEN1,2 변이유전자가 존재 시 응집되기 쉬운 끈적거리는 Aβ(1-42)를 과다하게 생성하여 독성 가용성 올리고머(OAβ)로 뭉치는 현상이 촉진되고 이로 인해 알츠하이머가 가속화된다.
PSEN1,2 변이유전자에 의한 독성 β-아밀로이드42 축적으로 Aβ(1-42):Aβ(1-40)의 비율이 50:1로 나타난다. 또한 건강한 뇌에서는 Aβ(1-42):Aβ(1-40)의 정상 비율이 1:9이다(그림 5). 이는 PSEN1,2 변이유전자가 존재 시 응집되기 쉬운 끈적거리는 Aβ(1-42)를 과다하게 생성하여 독성 가용성 올리고머(OAβ)로 뭉치는 현상이 촉진되고 이로 인해 발생되는 일련의 과정들에 의해 뉴런 사이의 시냅스 기능을 손상시킬 수 있음을 시사하며 이로 인해 알츠하이머가 가속화된다.
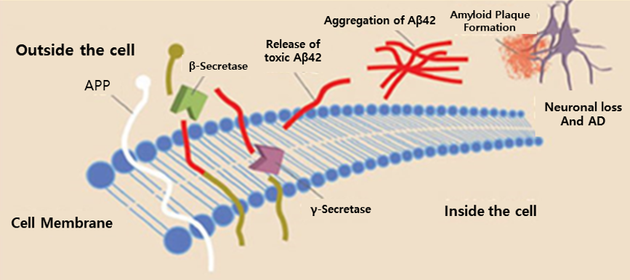
이 모든 유전자는 공통적으로 아밀로이드 생성에 대한 비정상적인 메커니즘을 가지고 있어 시간이 지남에 따라 계속되는 산화 스트레스와 생화학적 변화는 궁극적으로 알츠하이머의 전형적인 병리학적 과정인 신경섬유엉킴반(NeuroFibrillary Tangles, NFT)의 증가와 타우단백 응집에 이어 결국 신경세포 사멸로 이어진다(그림 6).
베클린1(Beclin1, BCEN1)은 알츠하이머 환자에서 감소하는 것으로 나타났으며, Beclin1의 결핍은 autophagosomal-lysosomal degradation을 정지시키고 Aβ 생성 및 Aβ plaque 축적을 증가시켜 질병을 가속화시킬 수 있다.
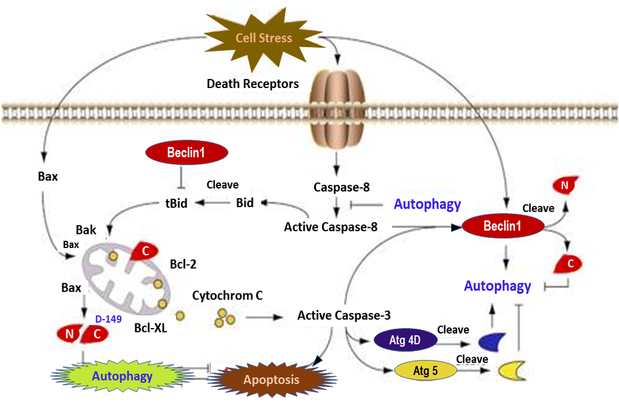
베클린1(Beclin1, BCEN1)은 오토파지(autophagy)의 개시와 조절에 관여하는 결정적인 단백질이며, 알츠하이머 환자에서 감소하는 것으로 나타났다. Autophagy는 그리스어로 “Self-Eating”의 의미를 담고 있으며, 단백질과 손상된 세포 기관을 포함한 세포질 구성 요소를 제거하고 세포재생을 위해 분해된 생체 분자를 재활용하는 세포 내 과정이다. 포유동물의 autophagy는 기아, 인슐린, 성장 인자, 저산소증 또는 병원균 감염 등과 같은 다양한 신호를 통해 촉진될 수 있다. 알츠하이머 환자의 뇌에서 Beclin1은 m-RNA와 단백질 모두에서 감소된다. 즉, 알츠하이머 환자의 경우 Beclin1의 결핍은 autophagosomal?lysosomal degradation을 정지시키고 Aβ 생성 및 Aβ plaque 축적을 증가시켜 질병을 가속화시킬 수 있다(그림 7).
Autophagy-lysosome 시스템의 기능 장애는 Aβ 축적과 타우 올리고머(tau oligomer) 및 NFT(neurofibrillary tangles)의 생성에 기여한다. 또한 자가탐식(autophagy)과 세포자살(apoptosis)은 공통적인 자극과 신호전달경로를 공유하고 어느 정도의 상호억제(crosstalk)를 나타낸다. Apoptotic stimuli에 지속적인 노출 시, Beclin1의 caspase-8 절단으로 N-terminal fragment와 C-terminal fragment를 생성하여 autophagy 기능을 잃게 한다. C-terminal fragment는 미토콘드리아로 전이되어 세포가 apoptotic signals에 민감하게 만들고 Beclin1과 Atg5 절단은 autophagy를 불활성화 시킨다. Caspase-3에 의한 Atg4D의 절단은 autophagy활성을 증가시키는 파편(fragment)을 생성한다. 더욱이, autophagy는 부분적으로 활성화된 caspase-8을 분해하거나 Beclin1에 의한 활성화를 막음으로써 apoptosis를 억제한다.
BeclinCCP항체(Beclin-1 caspase-cleavage product antibody)는 astrocyte의 건강마커이다. BeclinCCP 항체의 축적된 농도가 높으면 성상세포가 민감한 사멸경로를 겪고 있다는 것을 의미하며, 특히 알츠하이머 환자의 astrocyte에서 BeclinCCP 특이도가 높은 것으로 나타났다. 인간 아밀로이드 전구체 단백질(human Amyloid Precursor Protein, hAPP)을 발현하는 Beclin1 결핍 형질전환 마우스에서 Beclin1의 결핍을 채워 autophagy를 활성화시킨 결과 APP 및 대사산물의 농도가 감소되었다. 반면에 hAPP-발현 세포에서 autophagy를 억제한 결과 APP 및 대사산물의 축적을 의미하는 βCTF 농도가 높게 나타났다. Autophagy가 너무 많이 발생하거나 autophagic flux가 막히면 신경퇴화과정(neurodegeneration)으로 변할 수 있다. 실제로, autophagy의 퇴화가 알츠하이머 환자의 뇌에서 관찰되었으며, Beclin1의 부재 등과 같은 autophagy를 파괴시킨 APP형질 전환 마우스에서 이러한 autophagy의 파괴는 알츠하이머의 Aβ 병리를 악화시키며, 신경 퇴행을 촉진시키는 것이 관찰되었다.
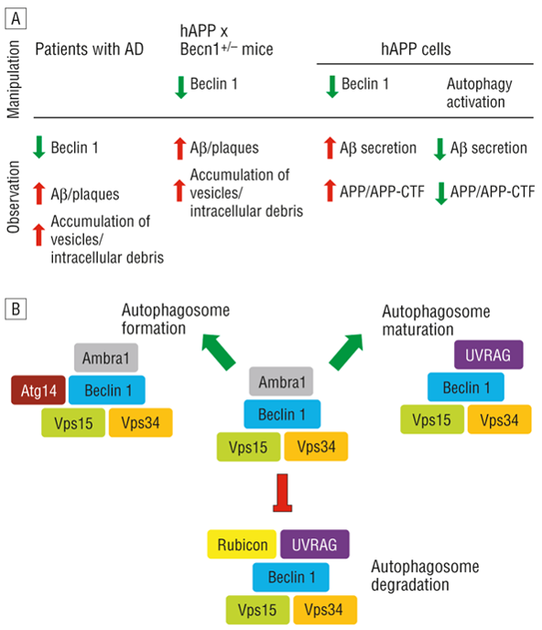
Beclin1은 다수의 결합 파트너를 가지고 있으며, Beclin1 복합체 내의 단백질 구성에 따라 그 기능을 결정하는 것으로 보인다. Beclin1의 농도감소는 복합체의 불안정화를 유발할 수 있으며 여러 단계에서 오토파지(autophagy)를 손상시킬 수 있다(그림 8). 또한 Beclin1-복합체의 조합은 세포나 조직에 따라 달라지며, Beclin1과의 상호 작용은 상대적으로 불안정하고 일시적이며 특정 질병 상황에서 다르게 발생할 수 있어서 autophagy의 자극과 억제 중 어느 것이 세포상황에 더 유익한지 여부를 결정하여 다양한 Beclin1-복합체로 발현하게 된다.
Autophagy는 수명이 긴 단백질과 세포 기관의 분해를 위한 주요 세포 경로이며 스트레스에 반응하여 세포의 운명을 조절하고 신경퇴행성 질환의 신경퇴화(neurodegeneration)와 연관이 있다. 그러나 autophagy의 역할이 알츠하이머에서 세포저하 및 세포유지 경로의 교란을 연결하는 해로운 역할인지 또는 보호하는 역할인지는 아직 불분명하며, 알츠하이머에서 autophagy의 역할은 세포 내 Aβ 응집체의 제거일 것으로 추정하고 있다.